Quantum-mechanical explanation of intermolecular interactions
In the natural sciences, an intermolecular force is an attraction between two molecules or atoms. They occur from either momentary interactions between molecules (the London dispersion force) or permanent electrostatic attractions between dipoles. They can be explained using a simple logical approach (see intermolecular force), or using a quantum mechanical approach.
Contents |
Perturbation theory
Hydrogen bonding, dipole–dipole interactions, and London (Van der Waals) forces are most naturally accounted for by Rayleigh–Schrödinger perturbation theory (RS-PT). In this theory—applied to two monomers A and B—one uses as unperturbed Hamiltonian the sum of two monomer Hamiltonians, 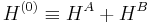
In the present case the unperturbed states are products 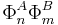 with
with 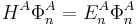 and
and 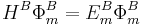
Supermolecular approach
The early theoretical work on intermolecular forces was invariably based on RS-PT and its antisymmetrized variants. However, since the beginning of the 1990s it has become possible to apply standard quantum chemical methods to pairs of molecules. This approach is referred to as the supermolecule method. In order to obtain reliable results one must include electronic correlation in the supermolecule method (without it dispersion is not accounted for at all), and take care of the basis set superposition error. This is the effect that the atomic orbital basis of one molecule improves the basis of the other. Since this improvement is distance dependent, it easily gives rise to artifacts.
Exchange
The monomer functions ΦnA and ΦmB are antisymmetric under permutation of electron coordinates (i.e., they satisfy the Pauli principle), but the product states are not antisymmetric under intermolecular exchange of the electrons. An obvious way to proceed would be to introduce the intermolecular antisymmetrizer 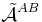 . But, as already noticed in 1930 by Eisenschitz and London,[1] this causes two major problems. In the first place the antisymmetrized unperturbed states are no longer eigenfunctions of H(0), which follows from the non-commutation
. But, as already noticed in 1930 by Eisenschitz and London,[1] this causes two major problems. In the first place the antisymmetrized unperturbed states are no longer eigenfunctions of H(0), which follows from the non-commutation
![\big[ \tilde{\mathcal{A}}^{AB}, H^{(0)}\big] \ne 0 .](/2012-wikipedia_en_all_nopic_01_2012/I/d29f9a88150d57a519f1ef3ccb864b89.png)
In the second place the projected excited states
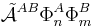
become linearly dependent and the choice of a linearly independent subset is not apparent. In the late 1960s the Eisenschitz–London approach was revived and different rigorous variants of symmetry adapted perturbation theory were developed. (The word symmetry refers here to permutational symmetry of electrons). The different approaches shared a major drawback: they were very difficult to apply in practice. Hence a somewhat less rigorous approach (weak symmetry forcing) was introduced: apply ordinary RS-PT and introduce the intermolecular antisymmetrizer at appropriate places in the RS-PT equations. This approach leads to feasible equations, and, when electronically correlated monomer functions are used, weak symmetry forcing is known to give reliable results.[2][3]
The first-order (most important) energy including exchange is in almost all symmetry-adapted perturbation theories given by the following expression
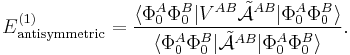
The main difference between covalent and non-covalent forces is the sign of this expression. In the case of chemical bonding this interaction is attractive (for certain electron-spin state, usually spin-singlet) and responsible for large bonding energies—on the order of a hundred kcal/mol. In the case of intermolecular forces between closed shell systems, the interaction is strongly repulsive and responsible for the "volume" of the molecule (see Van der Waals radius). Roughly speaking, the exchange interaction is proportional to the differential overlap between Φ0A and Φ0B. Since the wavefunctions decay exponentially as a function of distance, the exchange interaction does too. Hence the range of action is relatively short, which is why exchange interactions are referred to as short range interactions.
Electrostatic interactions
By definition the electrostatic interaction is given by the first-order Rayleigh–Schrödinger perturbation (RS-PT) energy (without exchange):
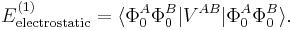
Let the clamped nucleus α on A have position vector Rα, then its charge times the Dirac delta function, Zα δ(r − Rα), is the charge density of this nucleus. The total charge density of monomer A is given by
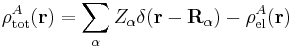
with the electronic charge density given by an integral over nA − 1 primed electron coordinates:
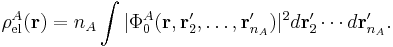
An analogous definition holds for the charge density of monomer B. It can be shown that the first-order quantum mechanical expression can be written as
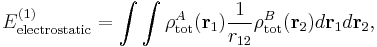
which is nothing but the classical expression for the electrostatic interaction between two charge distributions. This shows that the first-order RS-PT energy is indeed equal to the electrostatic interaction between A and B.
Multipole expansion
At present it is feasible to compute the electrostatic energy without any further approximations other than those applied in the computation of the monomer wavefunctions. In the past this was different and a further approximation was commonly introduced: VAB was expanded in a (truncated) series in inverse powers of the intermolecular distance R. This yields the multipole EXPANSION of the electrostatic energy. Since its concepts still pervade the theory of intermolecular forces, it will be presented here. In this article the following expansion is proved
![V^{AB} = \sum_{\ell_A=0}^\infty \sum_{\ell_B=0}^\infty (-1)^{\ell_B} \binom{2\ell_A%2B2\ell_B}{2\ell_A}^{1/2}
\sum_{M=-\ell_A-\ell_B}^{\ell_A%2B\ell_B} (-1)^{M} I_{\ell_A%2B\ell_B,-M}(\mathbf{R}_{AB})\; \left[\mathbf{Q}^{\ell_A} \otimes \mathbf{Q}^{\ell_B} \right]^{\ell_A%2B\ell_B}_M](/2012-wikipedia_en_all_nopic_01_2012/I/85f1f7b9b51ad528bf20a30080fee0bf.png)
with the Clebsch–Gordan series defined by
![\left[\mathbf{Q}^{\ell_A} \otimes \mathbf{Q}^{\ell_B} \right]^{\ell_A%2B\ell_B}_M \equiv
\sum_{m_A=-\ell_A}^{\ell_A} \sum_{m_B=-\ell_B}^{\ell_B}\;
Q_{m_A}^{\ell_A} Q_{m_B}^{\ell_B}\;\langle \ell_A, m_A; \ell_B, m_B| \ell_A%2B\ell_B, M \rangle.](/2012-wikipedia_en_all_nopic_01_2012/I/3c0e59d08ab63939395a56800583b411.png)
and the irregular solid harmonic is defined by
![I_{L,M}(\mathbf{R}_{AB}) \equiv \left[\frac{4\pi}{2L%2B1}\right]^{1/2}\;
\frac{Y_{L,M}(\widehat{\mathbf{R}}_{AB})}{R_{AB}^{L%2B1}}.](/2012-wikipedia_en_all_nopic_01_2012/I/36e036079607f7b79e200c90cc512318.png)
The function YL,M is a normalized spherical harmonic, while
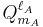 and
and 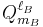 are spherical multipole moment operators. This expansion is manifestly in powers of 1/RAB.
are spherical multipole moment operators. This expansion is manifestly in powers of 1/RAB.
Insertion of this expansion into the first-order (without exchange) expression gives a very similar expansion for the electrostatic energy, because the matrix element factorizes,
![\begin{align}
E^{(1)}_\mathrm{electrostatic} = & \sum_{\ell_A=0}^\infty \sum_{\ell_B=0}^\infty (-1)^{\ell_B} \binom{2\ell_A%2B2\ell_B}{2\ell_A}^{1/2} \\
&\sum_{M=-\ell_A-\ell_B}^{\ell_A%2B\ell_B} (-1)^{M} I_{\ell_A%2B\ell_B,-M}(\mathbf{R}_{AB})\; \left[\mathbf{M}^{\ell_A} \otimes \mathbf{M}^{\ell_B} \right]^{\ell_A%2B\ell_B}_M,
\end{align}](/2012-wikipedia_en_all_nopic_01_2012/I/746ec3315e9317e677f9587002540afc.png)
with the permanent multipole moments defined by
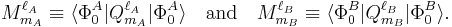
We see that the series is of infinite length, and, indeed, most molecules have an infinite number of non-vanishing multipoles. In the past, when computer calculations for the permanent moments were not yet feasible, it was common to truncate this series after the first non-vanishing term.
Which term is non-vanishing, depends very much on the symmetry of the molecules constituting the dimer. For instance, molecules with an inversion center such as a homonuclear diatomic (e.g., molecular nitrogen N2), or an organic molecule like ethene (C2H4) do not possess a permanent dipole moment (l=1), but do carry a quadrupole moment (l=2). Molecules such a hydrogen chloride (HCl) and water (H2O) lack an inversion center and hence do have a permanent dipole. So, the first non-vanishing electrostatic term in, e.g., the N2—H2O dimer, is the lA=2, lB=1 term. From the formula above follows that this term contains the irregular solid harmonic of order L = lA + lB = 3, which has an R−4 dependence. But in this dimer the quadrupole-quadrupole interaction (R−5) is not unimportant either, because the water molecule carries a non-vanishing quadrupole as well.
When computer calculations of permanent multipole moments of any order became possible, the matter of the convergence of the multipole series became urgent. It can be shown that, if the charge distributions of the two monomers overlap, the multipole expansion is formally divergent.
Ionic interactions
It is debatable whether ionic interactions are to be seen as intermolecular forces, some workers consider them rather as special kind of chemical bonding. The forces occur between charged atoms or molecules (ions). Ionic bonds are formed when the difference between the electron affinity of one monomer and the ionization potential of the other is so large that electron transfer from the one monomer to the other is energetically favorable. Since a transfer of an electron is never complete there is always a degree of covalent bonding.
Once the ions (of opposite sign) are formed, the interaction between them can be seen as a special case of multipolar attraction, with a 1/RAB distance dependence. Indeed, the ionic interaction is the electrostatic term with lA = 0 and lB = 0. Using that the irregular harmonics for L = 0 is simply
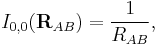
and that the monopole moments and their Clebsch–Gordan coupling are
![M^{0_A}_0 = q_A,\quad M^{0_B}_0 = q_B \quad\hbox{and}\quad [\mathbf{M}^{0_A}\otimes \mathbf{M}^{0_A}]^0_0 = q_A q_B ,](/2012-wikipedia_en_all_nopic_01_2012/I/2470d5dc366b298f5bdafc85f87e0703.png)
(where qA and qB are the charges of the molecular ions) we recover—as to be expected—Coulomb's law
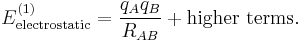
For shorter distances, where the charge distributions of the monomers overlap, the ions will repel each other because of inter-monomer exchange of the electrons.
Ionic compounds have high melting and boiling points due to the large amount of energy required to break the forces between the charged ions. When molten they are also good conductors of heat and electricity, due to the free or delocalized ions.
Writing
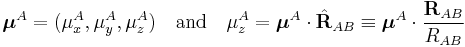
and similarly for B, we get the well-known expression
![E_{\mathrm{dip-dip}} = \frac{1}{R^{3}_{AB}}\left[ \boldsymbol{\mu}^A\cdot\boldsymbol{\mu}^B - 3 (\boldsymbol{\mu}^A\cdot \hat{\mathbf{R}}_{AB}) (\hat{\mathbf{R}}_{AB}\cdot \boldsymbol{\mu}^B) \right].](/2012-wikipedia_en_all_nopic_01_2012/I/e5fdf9dce22f63f05ed21bac182aab48.png)
As a numerical example we consider the HCl dimer depicted above. We assume that the left molecule is A and the right B, so that the z-axis is along the molecules and points to the right. Our (physical) convention of the dipole moment is such that it points from negative to positive charge. Note parenthetically that in organic chemistry the opposite convention is used. Since organic chemists hardly ever perform vector computations with dipoles, confusion hardly ever arises. In organic chemistry dipoles are mainly used as a measure of charge separation in a molecule. So,
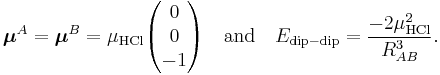
The value of μHCl is 0.43 (atomic units), so that at a distance of 10 bohr the dipole-dipole attraction is −3.698 10−4 hartree (−0.97 kJ/mol).
If one of the molecules is neutral and freely rotating, the total electrostatic interaction energy becomes zero. (For the dipole-dipole interaction this is most easily proved by integrating over the spherical polar angles of the dipole vector, while using the volume element sinθ dθdφ). In gases and liquids molecules are not rotating completely freely—the rotation is weighted by the Boltzmann factor exp(−Edip-dip/kT), where k is the Boltzmann constant and T the absolute temperature. It was first shown by John Lennard-Jones[4] that the temperature-averaged dipole-dipole interaction is
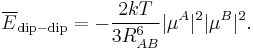
Since the averaged energy has an R−6 dependence, it is evidently much weaker than the unaveraged one, but it is not completely zero. It is attractive, because the Boltzmann weighting favors somewhat the attractive regions of space. In HCl-HCl we find for T = 300 K and RAB = 10 bohr the averaged attraction −62 J/mol, which shows a weakening of the interaction by a factor of about 16 due to thermal rotational motion.
Anisotropy and non-additivity of intermolecular forces
Consider the interaction between two electric point charges at position  and
and 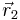 . By Coulomb's law the interaction potential depends only on the distance
. By Coulomb's law the interaction potential depends only on the distance 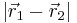 between the particles. For molecules this is different. If we see a molecule as a rigid 3-D body, it has 6 degrees of freedom (3 degrees for its orientation and 3 degrees for its position in R3). The interaction energy of two molecules (a dimer) in isotropic and homogeneous space is in general a function of 2×6−6=6 degrees of freedom (by the homogeneity of space the interaction does not depend on the position of the center of mass of the dimer, and by the isotropy of space the interaction does not depend on the orientation of the dimer). The analytic description of the interaction of two arbitrarily shaped rigid molecules requires therefore 6 parameters. (One often uses two Euler angles per molecule, plus a dihedral angle, plus the distance.) The fact that the intermolecular interaction depends on the orientation of the molecules is expressed by stating that the potential is anisotropic. Since point charges are by definition spherical symmetric, their interaction is isotropic. Especially in the older literature, intermolecular interactions are regularly assumed to be isotropic (e.g., the potential is described in Lennard-Jones form, which depends only on distance).
between the particles. For molecules this is different. If we see a molecule as a rigid 3-D body, it has 6 degrees of freedom (3 degrees for its orientation and 3 degrees for its position in R3). The interaction energy of two molecules (a dimer) in isotropic and homogeneous space is in general a function of 2×6−6=6 degrees of freedom (by the homogeneity of space the interaction does not depend on the position of the center of mass of the dimer, and by the isotropy of space the interaction does not depend on the orientation of the dimer). The analytic description of the interaction of two arbitrarily shaped rigid molecules requires therefore 6 parameters. (One often uses two Euler angles per molecule, plus a dihedral angle, plus the distance.) The fact that the intermolecular interaction depends on the orientation of the molecules is expressed by stating that the potential is anisotropic. Since point charges are by definition spherical symmetric, their interaction is isotropic. Especially in the older literature, intermolecular interactions are regularly assumed to be isotropic (e.g., the potential is described in Lennard-Jones form, which depends only on distance).
Consider three arbitrary point charges at distances r12, r13, and r23 apart. The total interaction U is additive; i.e., it is the sum
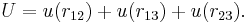
Again for molecules this can be different. Pretending that the interaction depends on distances only—but see above—the interaction of three molecules takes in general the form
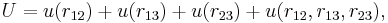
where 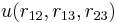 is a non-additive three-body interaction. Such an interaction can be caused by exchange interactions, by induction, and by dispersion (the Axilrod–Teller triple dipole effect).
is a non-additive three-body interaction. Such an interaction can be caused by exchange interactions, by induction, and by dispersion (the Axilrod–Teller triple dipole effect).
References
- ^ R. Eisenschitz and F. London, Z. Physik 60, 491 (1930) [1]. English translations in H. Hettema, Quantum Chemistry, Classic Scientific Papers, World Scientific, Singapore (2000), p. 336.
- ^ B. Jeziorski, R. Moszynski, and K. Szalewicz, Chem. Rev. 94, 1887–1930 (1994) [2].
- ^ K. Szalewicz and B. Jeziorski, in: Molecular Interactions, editor S. Scheiner, Wiley, Chichester (1995). ISBN 0471 959219.
- ^ J. E. Lennard-Jones, Proc. Phys. Soc. (London) 43, 461 (1931) [3].